")


CRISPR システム
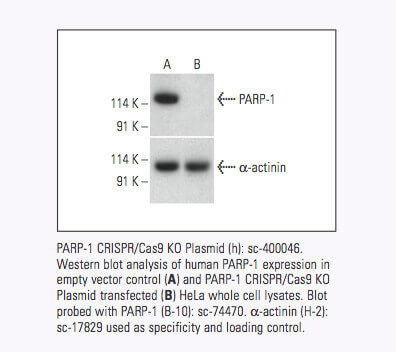
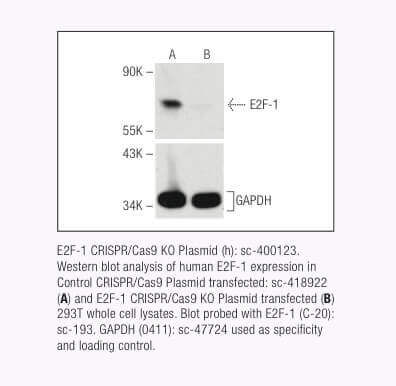
Santa Cruz Biotechnology社は現在、18,910以上のヒトおよび18,340以上のマウスのタンパク質コード遺伝子に対して、標的特異的CRISPR/Cas9ノックアウト(KO)プラスミド、CRISPRダブルニッカーゼプラスミド、CRISPR/ dCas9アクティベーションプラスミドおよびCRISPRレンチアクティベーションシステムを提供している。
Quick Links
CRISPR/Cas9の歴史
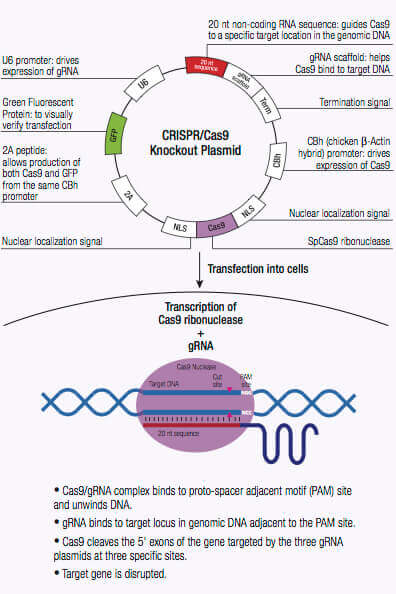
CRISPR/Casシステムは、古細菌や細菌における外来性遺伝物質の崩壊に利用される適応型免疫防御機序です。これらの微生物では、bacteriophageからの外来性遺伝物質が獲得され、CRISPR座位に組み込まれます (1,2)。spacerと知られるこの新物質は、将来のbacteriophage感染耐性に利用されるシークエンス特異的フラグメントが作られます。これらのシークエンス特異的断片は短鎖CRISPR RNAs(crRNAs)に翻訳され、CRISPR座位にエンコードされたCRISPR関連(Cas) 蛋白質のnuclease活性により、相補シークエンスをもつ侵入DNA断片を方向づけるガイドとし、機能します(1,2)。 II型CRISPRシステムのCas9 nucleaseにはRNA結合domain、alpha helix認識lobe(REC)、DNA切断用RuvCとHNHを含むnuclease lobe、およびprotospacer隣接motif(PAM)相互作用部位があります (1,2)。crRNAはREClobe内の架橋helixにの結合により、Cas9 nucleaseと複合体を形成し、crRNA骨格をもつ複数の塩橋を形成します (1,2,3)。
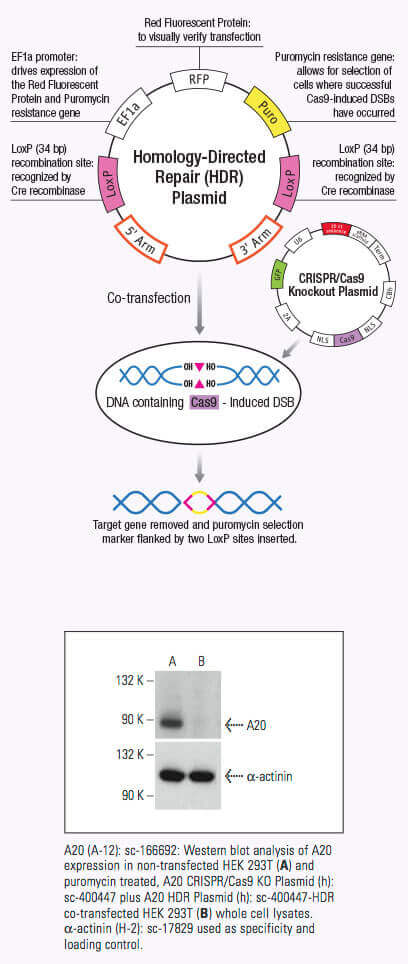
crRNAがCas9に結合すると、Cas9 nucleaseの立体構造が変化し、DNA結合用のchannelが作成されます(1,2,3)。Cas9/crRNA複合体はPAM(5’-NGG)部位(4,5,6)を探すため、DNAを走査します。PAM部位の認識はDNAの巻き戻しを誘導し、crRNAにPAM部位に隣接した相補鎖DNAを調べさせます。Cas9がcrRNAに相補なDNAシークエンスに隣接したPAM部位に結合すると、REC lobe内の架橋helixは、ターゲットDNAとともに、RNA-DNA heteroduplexが作られます (3,4,7)。PAM部位認識には核酸分解性HNHやRuvC domainの活性化が関与し、これによりターゲットDNAに二本鎖切断(DSB)が作られ、DNA崩壊を誘導します(1,2,5,8)。crRNAが相補的でない場合は、Cas9が放出され、次のPAM部位を検索します(7)。DNA内のターゲットゲノム鎖切断は非相同末端結合(NHEJ)修復経路により修復されますが、これにより挿入や欠失が導入され、エラーが生じます。ゲノム内の特異的部位でselected markersの組み換えに利用できる相同性配向型修復(HDR)経路により、修復されます (2,9,10)。このCRISPR/Cas9メカニズムは、哺乳動物細胞を含む多様なシステムにおけるゲノム工学に適用できます。
DSBs導入によるゲノム編集は、DNAシークエンスを認識するmeganucleases, zinc finger nucleases(ZNF)、またはtransactivator-like effectors (TALEs)を用い、実行されますが、それぞれには制限があります。meganuclesasesを使用する場合、nucleaseとDNAの間の部位特異的認識を明らかに示すことが困難です(2)。ほかの選択肢としたZNFやTALENでは3塩基未満のDNAシークエンスを設計し、認識することが困難であることが証明されています(2)。crRNAsのように作用する単一ガイドRNA(sgRNAs)は簡単に設計することができ、Cas9 nucleaseとともに同一vector内より発現させることにより、特異的なDNA部位をターゲットし、ゲノム編集することができます。CRISPR/Cas9システムは短鎖hairpin RNAsと比べ、感度も高く、screening目的では、効果がより高いです。
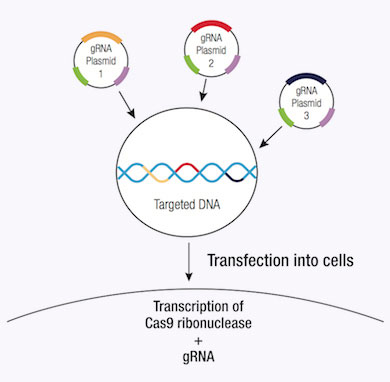
CRISPR/ Cas9システムによるゲノムDNAにDSBを誘導することの大きな利点は、その高いレベルの効率です。しかし、この効率は、特異性を低下させるオフターゲット効果の数により、ぼんやりする恐れがあるため、CRISPR/ Cas9編集の特異性を低下させます。特異性はCRISPRdouble nickaseシステムを使用することによって改善できるため、それぞれCas9(D10A)nickase変異体(Cas9n)をコードするペアのプラスミドは、標的特異的なガイドRNAによってゲノムDNA内の独特な、部位特定の領域に向けられます(12)。各Cas9n/ sgRNA複合体はガイドRNAに相補的なDNA鎖にnick一つのみを作成します(12)。ガイドRNAの各ペアは、約20 bpでずらし、標的DNAの反対鎖に位置する標的配列を認識します。Cas9n/ sgRNA複合体のペアによって作成されたdouble nickはDSBを模造します(12)。したがって、ペアリング?ガイドRNAの使用はCas9媒介遺伝子編集の増加した特異性を可能にしつつ、高いレベルの効率を維持します(12)。
ゲノム編集のほかに、CRISPRシステムは内因性遺伝子発現のロバストな活性化を可能にするように設計されました(13)。 CRISPRシステムのいくつかの構成要素は非常に効率的かつ特異的な転写活性化システムをもたらす相乗的活性化メディエーター(SAM)複合体を生成するように修飾されました(13)。 修正されたSAM複合体の構成要素の一つは、Cas9 nucleaseです。SAMシステムで は、Cas9の触媒ドメインが非活性化され、得られたdCas9は転写活性化ドメイン (VP64)に融合されます。標的特異的なガイドRNA (sgRNA)によって指示され、 dCas9-VP64-sgRNA複合体は遺伝子発現を上方制御する内在性遺伝子の転写開始部位(TSS)から-200 bpの領域を標的とします(13)。転写をさらに増強するため、sgRNAはtetraloopとstem loop 2に最小のヘアピンアプタマーを追加することにより、修正されました(13)。sgRNAにあるアプタマーは、二量体化されたMS2バクテリオファージのコート蛋白質を選択的に結合します (13)。p65とHSF1トランス活性化ドメインにMS2蛋白質を融合させることは得られたMS2-P65-HSF1融合蛋白質を転写因子のリクルートメントを強化させることを可能にします。それによって、dCas9媒介された遺伝子活性化の効力を向上させます (13)。すべての細胞型にSAMの転写活性化システムを効率的に引渡すため、活性化製品はトランスフェクション用の標準プラスミドならびにレンチウイルス包装と形質導入用のレンチウイルスプラスミドで提供します (13)。
Quick Links
CRISPR/Cas9主導の二本鎖切断(DSB)