")


CRISPR Systems
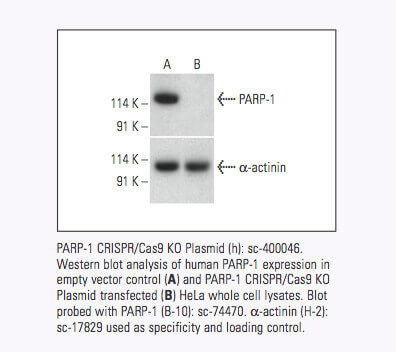
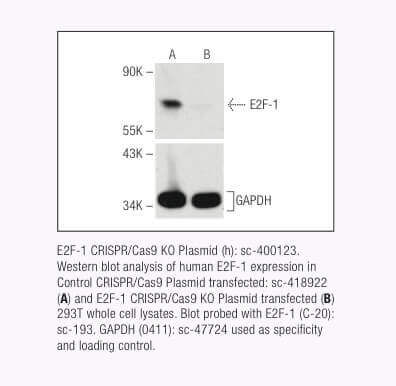
Santa Cruz Biotechnology now offers target-specific CRISPR/Cas9 Knockout (KO) Plasmids, CRISPR Double Nickase Plasmids, CRISPR/ dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for over 18,910 human and 18,340 mouse protein encoding genes.
Quick Links
History of CRISPR/Cas9
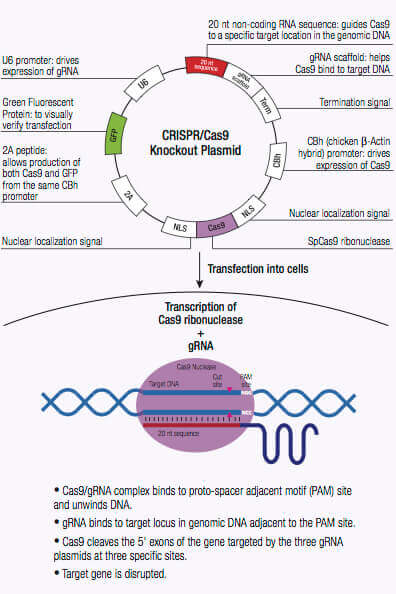
The CRISPR/Cas system is an adaptive immune defense mechanism used by Archea and bacteria for the degradation of foreign genetic material. In these organisms, the foreign genetic material from a bacteriophage is acquired and integrated into the CRISPR loci (1,2). This new material, also known as a spacer, creates a sequence-specific fragment used for future resistance against a bacteriophage infection. These sequence-specific fragments are translated into short CRISPR RNAs (crRNAs) and function as a guide to direct cleavage of complementary invading DNA via the nuclease activity of CRISPR-associated (Cas) protein also encoded by the CRISPR loci (1,2). Cas9 nuclease of the type II CRISPR system has an RNA binding domain, an alpha helix recognition lobe (REC), a nuclease lobe that include the RuvC and HNH for DNA cleavage, and a protospacer adjacent motif (PAM) interacting site (1,2). crRNA forms a complex with the Cas9 nuclease by binding to the bridge helix within the REC lobe, and forms multiple salt bridges with the backbone of the crRNA (1,2,3).
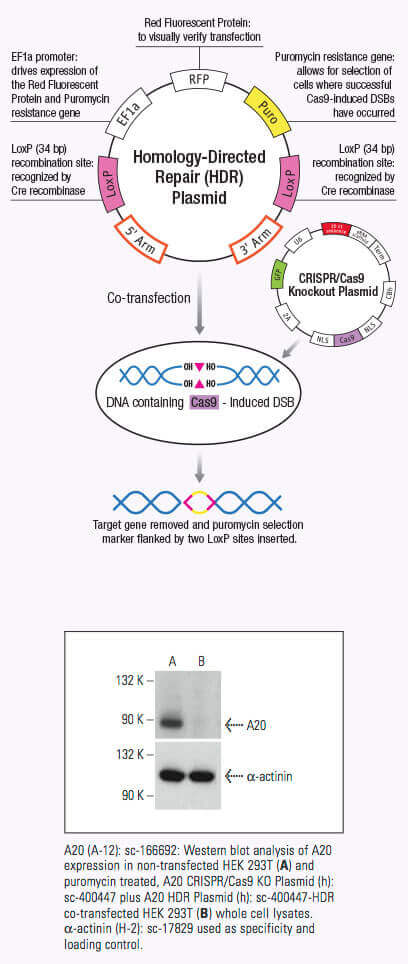
Once the crRNA binds to the Cas9 the conformation of the Cas9 nuclease changes and creates a channel that allows for DNA binding (1,2,3). The Cas9/crRNA complex scans the DNA for a PAM (5'-NGG) site (4,5,6). Recognition of a PAM site leads to unwinding of the DNA, and allows the crRNA to check for complementary DNA adjacent to the PAM site. When Cas9 binds to a PAM site adjacent to a DNA sequence that is complementary to the crRNA, the bridge helix within the REC lobe creates an RNA-DNA heteroduplex with the target DNA (3,4,7). The PAM site recognition is involved in activating the nucleolytic HNH and RuvC domains which create a double-stranded break (DSB) in the target DNA, leading to DNA degradation (1,2,5,8). If the crRNA is not complementary, then Cas9 releases and searches for another PAM site (7). Targeted genome strand breaks in the DNA can be repaired via the nonhomologous end-joining (NHEJ) repair pathway, which introduces insertion or deletions creating errors, or through the homologous directed repair (HDR) pathway, that can be used to recombine selected markers at specific sites in the genome (2,9,10). This CRISPR/Cas9 mechanism can be repurposed for genomic engineering of various systems, including mammalian cells.
Genome editing via introduction of DSBs can be performed with meganucleases, zinc finger nucleases (ZF), or transactivator-like effectors (TALEs), which recognize DNA sequences, however, each has their limitations. When using meganuclesases it is difficult to clearly show site-specific recognition between nuclease and DNA (2).The other options, ZFs and TALEs, have proven difficult to design and recognize up to 3 nt of DNA (2). Single guide RNAs (sgRNAs) that act like crRNAs are easily designed and can be expressed along with Cas9 nuclease in the same vector to target specific DNA sites for genome editing. The CRISPR/Cas 9 system also has higher sensitivity and is more efficient when used for screening than small hairpin RNAs.
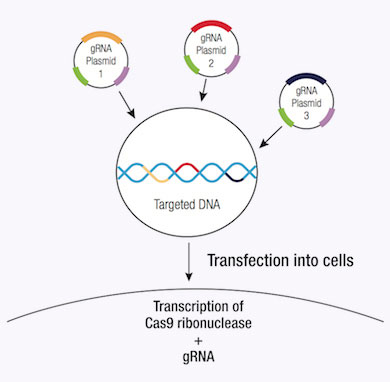
A significant advantage of using the CRISPR/Cas9 system to induce DSB in genomic DNA is its high level of efficiency. However, this efficiency can be clouded by a number of off-target effects, thereby reducing the specificity of CRISPR/Cas9 editing. Specificity can be improved by using a CRISPR double nickase system, whereby a pair of plasmids, each encoding a Cas9 (D10A) nickase mutant (Cas9n) are directed to a distinct, site specific region in the genomic DNA by a target-specific guide RNA (12). Each Cas9n/sgRNA complex creates only one nick in the DNA strand that is complementary to the guide RNA (12). Each pair of guide RNAs are offset by approximately 20 bp and recognize target sequences located on opposite strands of the target DNA. The double nick created by the pair of Cas9n/sgRNA complexes mimics a DSB (12). Thus, the use of paired-guide RNAs allows for increased specificity of Cas9-mediated gene editing, while maintaining a high level of efficiency (12).
In addition to genome editing, the CRISPR system has been engineered to allow for robust activation of endogenous gene expression (13). Several components of the CRISPR system have been modified to generate the synergistic activation mediator (SAM) complex that results in a highly efficient and specific transcription activation system (13). One component of the SAM complex that is modified is the Cas9 nuclease. In the SAM system, the catalytic domains of Cas9 have been deactivated and the resulting dCas9 is fused to a transcription activation domain (VP64). Directed by a target specific guide RNA (sgRNA), the dCas9-VP64-sgRNA complex targets the -200 bp region from the Transcriptional Start Site (TSS) of endogenous genes to upregulate gene expression (13). To further enhance transcription, the sgRNA has been modified by appending a minimal hairpin aptamer to the tetraloop and stem loop 2 (13). This aptamer on the sgRNA selectively binds dimerized MS2 bacteriophage coat proteins (13). Fusing the MS2 proteins to p65 and HSF1 transactivation domains allows the resulting MS2-P65-HSF1 fusion protein to enhance the recruitment of transcription factors, thereby improving the potency of dCas9-mediated gene activation (13). Activation products are supplied as both standard plasmids for transfection and lentiviral plasmids for lentiviral packaging and transduction, for efficient delivery of the SAM Transcription Activation System into all cell types (13).
Quick Links
CRISPR/Cas9-directed Double Strand Break (DSB)