")
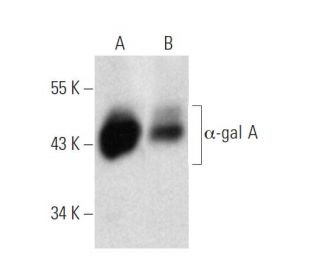
: sc-517442. Analyse par Western blot de l'expression de l'α-gal A dans des lysats de cellules entières 293T (A) et NCI-H460 (B).")
Anticorps α-gal A (C6): sc-517442
- L'anticorps alpha-gal A C6 est un monoclonal IgG2b (chaîne légère kappa) L'Anticorps α-gal A fourni en 100 µg/ml
- élevé contre une protéine recombinante correspondant aux acides aminés 81-429 de α-gal A d'origine human.
- recommandé pour la détection de α-gal A d'origine human par WB, IP, IF, IHC(P) et ELISA
- m-IgG Fc BP-HRP et 2b BP-HRP">m-IgG2b BP-HRP sont les réactifs de détection secondaire préférés pour α-gal A Antibody (C6) pour les applications WB et IHC(P). Ces réactifs sont désormais proposés en lots avec α-gal A Antibody (C6)(voir les informations de commande ci-dessous).

ACCÈS RAPIDE AUX LIENS
VOIR ÉGALEMENT...
L'anticorps α-gal A (C6) est un anticorps monoclonal IgG2b à chaîne légère kappa de souris qui détecte la protéine alpha-gal A d'origine humaine par western blotting (WB), immunoprécipitation (IP), immunofluorescence (IF), immunohistochimie sur coupes incluses en paraffine (IHCP) et dosage immunoenzymatique (ELISA). L'anticorps alpha-gal A (C6) est disponible sous forme d'anticorps non conjugué. La protéine alpha-galactosidase A (α-gal A) joue un rôle crucial en tant qu'hydrolase lysosomale, en décomposant les glycolipides, en particulier le globotriaosylcéramide (Gb3). Cette activité enzymatique est vitale pour le maintien de l'homéostasie cellulaire, car l'accumulation de Gb3 due à des déficiences en α-gal A peut entraîner de graves problèmes de santé, notamment la maladie de Fabry, une maladie récessive liée au chromosome X caractérisée par l'accumulation de glycolipides dans divers tissus. Chez les patients atteints de la maladie de Fabry, l'absence d'α-gal A fonctionnelle entraîne des complications rénales et cardiaques, de fortes douleurs dans les extrémités et des lésions cutanées caractéristiques appelées angiokératomes. La thérapie de remplacement enzymatique utilisant l'α-gal A recombinante s'est avérée efficace pour soulager les symptômes associés à cette maladie débilitante, soulignant l'importance de l'α-gal A dans les interventions thérapeutiques.
Alexa Fluor® est une marque déposée de Molecular Probes Inc., OR., USA
LI-COR® et Odyssey® sont marques déposées de LI-COR Biosciences
α-gal A Références:
- La perfusion d'alpha-galactosidase A réduit le stockage de globotriaosylcéramide dans les tissus chez les patients atteints de la maladie de Fabry. | Schiffmann, R., et al. 2000. Proc Natl Acad Sci U S A. 97: 365-70. PMID: 10618424
- Maladie de Fabry: des études précliniques démontrent l'efficacité du remplacement de l'alpha-galactosidase A chez des souris déficientes en enzyme. | Ioannou, YA., et al. 2001. Am J Hum Genet. 68: 14-25. PMID: 11115376
- Essai clinique de phase 1/2 de remplacement enzymatique dans la maladie de Fabry: études de pharmacocinétique, de clairance du substrat et de sécurité. | Eng, CM., et al. 2001. Am J Hum Genet. 68: 711-22. PMID: 11179018
- Maladie de Fabry: diagnostic et traitement. | Breunig, F., et al. 2003. Kidney Int Suppl. S181-5. PMID: 12694340
- Une approche pharmacogénétique pour identifier les formes mutantes de l'alpha-galactosidase A qui répondent à une chaperonne pharmacologique pour la maladie de Fabry. | Wu, X., et al. 2011. Hum Mutat. 32: 965-77. PMID: 21598360
- Caractérisation fonctionnelle des mutations de l'alpha-galactosidase A comme base d'un nouveau système de classification de la maladie de Fabry. | Lukas, J., et al. 2013. PLoS Genet. 9: e1003632. PMID: 23935525
- Conséquences fonctionnelles et cliniques des nouvelles mutations de l'alpha-galactosidase A dans la maladie de Fabry. | Lukas, J., et al. 2016. Hum Mutat. 37: 43-51. PMID: 26415523
- Le génotype N215S de l'&alpha-galactosidase A induit une variante cardiaque spécifique de la maladie de Fabry. | Oder, D., et al. 2017. Circ Cardiovasc Genet. 10: PMID: 29018006
- Activité de l'alpha-galactosidase A dans la maladie de Parkinson. | Alcalay, RN., et al. 2018. Neurobiol Dis. 112: 85-90. PMID: 29369793
- Le rapport alpha-galactosidase A/lysoGb3 comme marqueur potentiel de la maladie de Fabry chez les femmes. | Baydakova, GV., et al. 2020. Clin Chim Acta. 501: 27-32. PMID: 31770509
- Maladie de Fabry: déficit en alpha-galactosidase. | Kint, JA. 1970. Science. 167: 1268-9. PMID: 5411915
- Cartographie physique dans la région des loci des gènes de la tyrosine kinase de Bruton et de l'alpha-galactosidase A dans la partie proximale de Xq22. | Sweatman, AK., et al. 1994. Hum Genet. 94: 624-8. PMID: 7989038
Informations pour la commande
| Nom du produit | Ref. Catalogue | COND. | Prix HT | QTÉ | Favoris | |
Anticorps α-gal A (C6) | sc-517442 | 100 µg/ml | $322.00 | |||
α-gal A (C6): m-IgG Fc BP-HRP Kit | sc-541107 | 100 µg Ab; 10 µg BP | $361.00 | |||
α-gal A (C6): m-IgG2b BP-HRP Kit | sc-549805 | 100 µg Ab; 10 µg BP | $361.00 |